
CQA(주요 품질 특성)
원하는 제품 품질을 보장하기 위해 적절한 한도, 범위, 분포 이내에 있어야 하는 물리적, 화학적, 생물학적, 미생물학적 특성
-ICH Q8(R2)
환자와 임상 관점인 QTPP와 달리 CQA는 의약품 개발자의 관점에서 중요한 품질 특성을 의미함
QTPP 항목들 중 제제(Formulation)나 공정 변수에 의해서 변경될 가능성이 높은 품질 항목
CQA의 선정 절차

1. 잠재 QA 도출
모든 의약품 품질 특성을 도출
물리적 특성, 확인, 함량, 함량균일성, 분해산물
잔류용매, 수분, 미생물한도 등
2. Criticality 평가
환자에게 끼치는 해의 심각성 평가
QA(Quality Attribute, 품질 특성) 요구사항을
충족시키지 못했을 때 안정성과 유효성의 측면
3. CQA 선정
위험 관리를 고려하기 전 평가해서 선정
관리가 잘 된다고 해서 심각성이 낮은 것이 아님
제네릭 의약품의 CQA 작성 사례
1. 경구 투여 정제
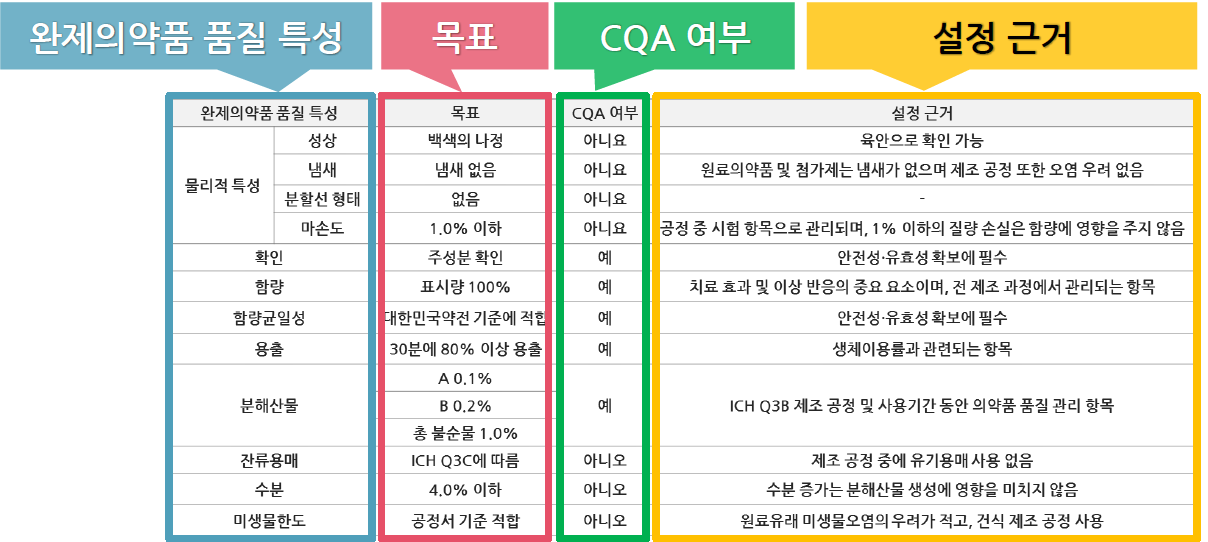
|
완제의약품 품질 특성 |
목표 |
CQA 여부 |
설정 근거 |
|
|
물리적 특성 |
성상 |
백색의 나정 |
아니요 |
육안으로 확인 가능 |
|
냄새 |
냄새 없음 |
아니요 |
원료의약품 및 첨가제는 냄새가 없으며 제조 공정 또한 오염 우려 없음 |
|
|
분할선 형태 |
없음 |
아니요 |
- |
|
|
마손도 |
1.0% 이하 |
아니요 |
공정 중 시험 항목으로 관리되며, 1% 이하의 질량 손실은 함량에 영향을 주지 않음 |
|
|
확인 |
주성분 확인 |
예 |
안전성·유효성 확보에 필수 |
|
함량 |
표시량 100% |
예 |
치료 효과 및 이상 반응의 중요 요소이며, 전 제조 과정에서 관리되는 항목 |
|
함량균일성 |
대한민국약전 기준에 적합 |
예 |
안전성·유효성 확보에 필수 |
|
용출 |
30분에 80% 이상 용출 |
예 |
생체이용률과 관련되는 항목 |
|
분해산물 |
A 0.1% |
예 |
ICH Q3B 제조 공정 및 사용기간 동안 의약품 품질 관리 항목 |
|
B 0.2% |
|||
|
총 불순물 1.0% |
|||
|
잔류용매 |
ICH Q3C에 따름 |
아니오 |
제조 공정 중에 유기용매 사용 없음 |
|
수분 |
4.0% 이하 |
아니오 |
수분 증가는 분해산물 생성에 영향을 미치지 않음 |
|
미생물한도 |
공정서 기준 적합 |
아니오 |
원료유래 미생물오염의 우려가 적고, 건식 제조 공정 사용 |
바이오 의약품의 CQA 선정
바이오 의약품의 CQA도 합성의약품과 같은 절차로 선정
1. Criticality 분석을 위한 도구
1) FMEA(Failure Modes & Effects Analysis)
2) FMECA(Failure Modes, Effects and Criticality Analysis) : 고장 모드, 효과 및 임계 분석
① 프로세스의 잠재적 위험 요소를 규명하고 제품 성능에 미치는 영향을 평가하는 도구
② 제품 및 프로세스에 대한 이해를 바탕으로 중요한 위험 요소, 위험을 야기하는 요인 및 영향을 파악하는 데 활용
③ 결과의 심각도, 발생 가능성 및 검출 가능성 조사
- 위험의 우선 순위를 정함
- 위험 통제 활동의 효과를 모니터링하는 데 사용
④ FMEA의 결과에 중요성 분석을 추가해 분석하는 도구
⑤ 제약 산업에서 주로 제조 공정과 관련된 실패 및 위험 평가나 위험 순위 평가에 활용
⑥ FMECA의 결과물은 각 위험 모드에 대한 상대적 위험 점수
- 상대적 위험을 기준으로 위험 요소의 순위를 매김
3) FTA(Fault Tree Analysis) : 오류 트리 분석
① 제품 또는 프로세스의 기능 실패에 대해 한 번에 하나씩 인과 관계를 식별한 후, 이들을 결합하여 트리 구조로 표시하는 방법
② 트리의 각 레벨에서 실패 모드의 조합을 AND, OR 등의 논리 연산자로 설명
③ 실패의 근본 원인에 대한 경로를 파악하여 여러 요소가 주어진 문제에 어떻게 영향을 미치는지 평가하는 효과적인 도구
④ 실패 원인 요소의 개선이 다른 문제를 발생시키지 않고 해당 문제를 해결할 수 있는지 확인
4) HACCP(Hazard Analysis and Critical Control Points)
① 제품 품질, 신뢰성 및 안전성을 보장하기 위한 체계적·예방적인 도구
② 제품의 설계, 개발, 생산 및 사용 중에 발생하는 위험 요소와 영향을 분석, 평가, 방지, 제어하는 과학적·체계적 방식
③ 7단계로 구성됨
- 유해성 분석을 실시하고 공정의 각 단계에 대한 예방 조치 확인
- 임계점 결정
- 임계치 설정
- 감시 시스템 수립
- 모니터링의 중요 통제점이 제어 상태가 아님을 나타낼 때 수행할 시정 조치 수립
- HACCP 시스템이 효과적으로 작동하는지 확인하기 위한 시스템 수립
- 기록 보관 시스템 수립
④ 물리적·화학적·생물학적 위험과 관련된 위험을 식별하고 관리하는 데 사용
⑤ 중요 통제점을 식별할 정도로 제품 및 프로세스에 대한 이해가 충분할 때 가장 유용
⑥ HACCP 분석 결과를 활용하여 제조 프로세스뿐만 아니라 전체 라이프 사이클 단계도 쉽게 모니터링
5) HAZOP(Hazard Operability Analysis)
① 위험 상황이 의도된 설계 또는 작동으로부터의 편차에 기인한다고 가정하여 만들어진 도구
② ‘Guide-word’를 사용하여 위험을 식별하는 체계적인 브레인 스토밍 기법
- Guide-word : No, More, Other Than, Part of 등의 단어
③ 업스트림 공급 업체,장비 및 시설뿐만 아니라 제조 프로세스 전체에 적용
④ HAZOP 분석 결과는 리스크 관리 운영 목록으로 제조 프로세스의 중요 포인트를 정기적으로 모니터링할 수 있음
6) PHA(Preliminary Hazard Analysis)
① 사전 경험을 바탕으로 미래의 위험 요소를 식별하고 발생 확률 분석
② 광범위한 기법을 사용하지 못하는 환경에서 기존 시스템 분석 및 위험의 우선 순위 정하는 경우 유용
③ 설계 세부 사항이나 운영 절차에 대한 정보가 거의 없는 프로젝트 개발 초기에 사용
④ PHA 구성 요소
- 위험 이벤트가 발생할 수 있는 가능성의 확인
- 발생할 수 있는 상해 또는 손상의 정도에 대한 정성적 평가
- 심각성과 발생도를 조합한 위험도의 상대적 순위 가능한 조치 방법의 확인
7) RRF(Risk Ranking and Filtering) : 바이오 의약품의 경우 주로 RRF를 사용함
① 위험을 비교하고 순위를 매기는 도구
② 다양한 양적, 질적 요소들에 대한 평가 점수를 하나의 상대 위험 점수로 통합하여
③ 위험도 순위 지정
④ 커트라인 개념인 ‘필터’를 사용하여 위험 순위를 관리 또는 조정
⑤ 제조 현장의 우선 순위를 정함
⑥ 위험 요소 또는 결과가 다양하여 한가지 도구로
⑦ 비교하기 어려운 경우 유용
⑧ 동일 조직 프레임 워크 내에서 정량적으로 평가된 위험과 정성적으로 평가된 위험을 모두 평가할 때 유용
RRF(위험 순위 매김)
Criticality Score = Impact X Uncertainty
1. Impact(영향도)
Efficacy, PK/PD, Immunogenicity, Safety의 4가지 항목에 영향을 주는 정도를 평가
2. Uncertainty(불확실도)
Impact 판단을 위해 사용한 정보의 출처를 평가
3. Score 값이 기준값을 초과하면 CQA로 판정, 기준값 이하이면 Non-CQA로 판정
4. 일반적으로 기준값은 20(A-Mab Case 참조)
Impact 평가 기준 예: A-Mab Case
|
Impact (Score) |
Biological Activity or Efficacya |
PK/PDa |
Immunogenicity |
Safety |
|
Very High (20) |
Very significant change |
Significant change on PK |
ATA detected and confers limits on safety |
Irreversible AEs |
|
High (16) |
Significant change |
Moderate change with impact on PD |
ATA detected and confers limits on efficacy |
Reversible AEs |
|
Moderate (12) |
Moderate change |
Moderate change with no impact on PD |
ATA detected with in vivo effect that can be managed |
Manageable AEs |
|
Low (4) |
Acceptable change |
Acceptable change with no impact on PD |
ATA detected with minimal in vivo effect |
Minor, transient AEs |
|
None (2) |
No change |
No impact on PK or PD |
ATA not detected or ATA detected with no relevant in vivo effect |
No AEs |
Uncertainty 평가 기준 예: A-Mab Case
|
Uncertainty (Score) |
Description (Variants and Host Related Impurities) |
Description (Process Raw Material) a |
|
7(Very High) |
No information (new variant) |
No information (new impurity) |
|
5(High) |
Published external literature for variant in related molecule. |
--- |
|
3(Moderate) |
Nonclinical or in vitro data with this molecule. Data (nonclinical, in vitro or clinical) from a similar class of molecule. |
Component used in previous processes |
|
2(Low) |
Variant has been present in material used in clinical trials. |
--- |
|
1(Very Low) |
Impact of specific variant established in Clinical Studies with this molecule. |
GRAS or studied in clinical trials |
GRAS = generally regarded as safe
a Assesses the impact of a raw material as an impurity. Impact of the raw material on the product during manufacturing is assessed during process development.
CQA Criticality 평가 사례 : A-Mab Case
|
Tool #1 (Impact × Uncertainty) |
|||||
|
Attribute |
Efficacy |
PK/PD |
Immunogenicity |
Safety |
Risk Score |
|
Galactose Content |
16 × 3 = 48 |
2 × 5 = 10 |
2 × 5 = 10 |
2 × 5 = 10 |
48 |
Risk Score : 4개 항목 평가 점수 중 최대값인 48점
Risk Score 값이 20을 초과하므로 CQA로 판정
CQA-AC(Acceptance Criteria, 허용 기준) 설정
의약품의 안정성과 유효성을 보장하기 위해서 CQA가 만족해야 하는 허용 범위
CPP(주요 공정 인자) 허용 범위 설정에 필요
→ 개발 초기, 정보 부족으로 인해 임시로 설정 후, Process Characterization 단계 이전에 확정
1. AC 설정방법
1) 사전 지식(Prior Knowledge) : 관련 문헌, 타사/자사 유사 제품
2) In-vitro 연구 결과
3) 비임상 연구 결과
4) 임상 연구 결과와 상업 생산 데이터 등 활용

댓글