
HF(Hartree-Fock) 이론과 DFT(Density Functional Theory)를 포함한 분자설계과정의 기본이론을 이해하고, Gaussian09W 프로그램을 이용하여 분자 모델링의 기초를 숙지한다.
실험에서 사용하는 시약인 Alq3(Tris(8-hydroxyquinolinato)aluminium) 에 대한 분자 모형을 설계하고, 프로그램을 실행하여 그 결과를 통해 Alq3의 안정된 구조와 에너지, IR 스펙트럼 등의 정보를 얻을 수 있다.
Computational Chemistry
계산 화학은 분자, 원자, 원자의 구성 입자의 운동과 상태를 나타내는 함수를 컴퓨터를 이용해 계산함으로써 화학 물질들의 이론적인 문제를 다루는 학문이다. 이번 실험 시간에는 Gaussian09W 프로그램을 사용하여 물리학적 법칙에 들어맞는 분자 구조를 그린 후(computational model), 여러 조작을 통해 그 분자의 운동 상태, 분자궤도함수(molecular orbital), 분자 구조에 따른 에너지 변화 등에 대한 정보를 얻고 에너지가 가장 낮은 상태로 최적화 시키는 과정을 수행하였다.
여기서 더 나아가 분자 내 원자의 움직임을 파악하여 다른 물리화학적 성질을 계산해낼 수 있으며, 실험적인 관찰만으로는 얻기 힘들었던 불안정한 분자(중간 생성물, 전이 상태 분자 등)의 물리적 정보(에너지, 쌍극자 모멘트, 열역학적 정보 등)와 분자 사이의 반응에 대한 정보도 얻을 수 있다. 계산 화학은 여러 가지 방법(method)으로 수행된다. 방법들은 크게 두 가지 방법으로 나뉘어 지는데 고전 역학에 기반을 둔 분자역학과 양자역학에 기반을 둔 전자 구조 방법(Electronic Structure Method)이다.
실험 방법
Step 1
Gaussian 09(혹은 ver. 03)이라는 프로그램으로 기본적인 계산을 하는데 사용법은 생략하고 아래의 그림의 Job Entry를 보면서 설명을 하면 5곳의 입력란이 있는데 %Section을 생략하고 첫째 Route Section부터 보겠다.
Route Section은 이론적인 모델과 계산형태를 적는곳이고, 둘째 Title Section은 간단한 설명을 적는 곳이고, 셋째 Charge, Multipl은 전하와 다중도를 적고, 넷째 Molecular Specification은 분자의 구조를 적는다.
그래서 실험을 수행하는데 계산하는 방법은 최적화에(Optimized) 대한 Frequency로 하여 제시한 분자에 대한 계산을 하는데 순서는 Input파일을 만드는 것에서부터 있다.
Input파일은 semi-empirical(반경험적인) 중에 pm3 methods를 사용하였다.
Route section 부분에 입력하는 사항은 #t PM3 Opt Freq로 하고 Charge, Multiple는 0과 1을 주어 입력값을 넣고 분자구조인 Z-matrix를 짜서 입력한다
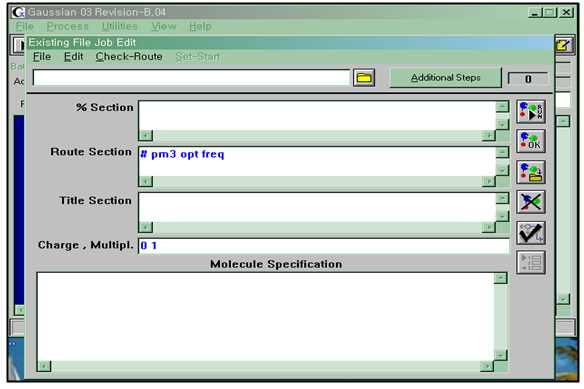
Step 2
결과물을 출력하기 위한 Output파일을 생성해야만 계산이 실행하게 된다. 이때 Scratch폴더에 자동으로 chk파일이 만들어진다.
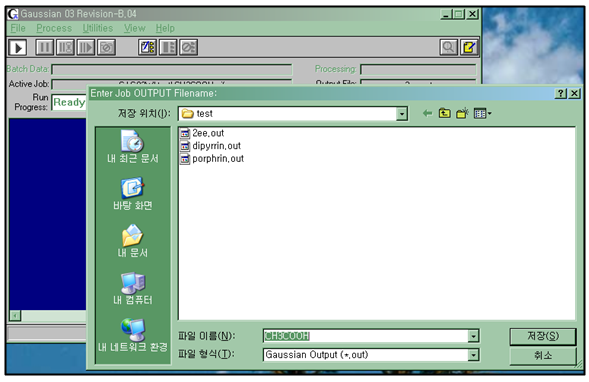
Step 3
Output파일을 만들고 계산이 실행되는 과정을 지켜보면서 Semi-empirical 방법적으로 계산이 되는지 확인을 하고 끝마치면 최적화 된 Energy 값을 찾아 분석을 한다.

[물리화학실험]Molecular modeling - Gaussian03W calculation study 레포트
1.1. Computational Chemistry 계산 화학은 분자, 원자, 원자의 구성 입자의 운동과 상태를 나타내는 함수를 컴퓨터를 이용해 계산함으로써 화학 물질들의 이론적인 문제를 다루는 학문이다. 이번 실험 �
www.happycampus.com

댓글