
Gene cloning으로 DNA를 자르고 붙이고 변형시켜서 실험에 필요한 재조합 DNA를 제조하고, 기본적인 효소의 처리와 DNA의 분리 및 조작방법 이용하여 분자생물학의 기본을 배우기 위함이다.
Gene cloning
동식물의 한 개체에서 수정을 거치지 않고, 무성생식에 의하여 양친과 똑같은 유전자 조성을 가진 개체를 얻는 기술이다. 그 개체군을 클론이라고 하며, 꺾꽃이, 접붙이기, 포기나누기 등으로 증식시킨 개체도 클론이라고 일컫는다. 클로닝은 암수의 유전자가 서로 섞이지 않으므로 우수한 품종을 보존하기 위한 가장 좋은 방법이다.
당근, 담배 등과 같은 식물에서는 성공사례가 많으며, 육종에 효과적으로 이용하고 있다. 동물의 경우도 양서류에서 성공하고 있다. 1963년 J.B 가든은 아프리카 개구리의 체세포에서 핵을 꺼내어, 그것을 핵을 빼낸 알에 이식해서 새로운 개체를 만드는 데 성공하였다. 포유류에서는 스위스의 K.이르멘제 등이 흰쥐를 사용해서 실험에 성공하였다. 수정한 지 얼마 안 되는 태아기 세포로부터 핵을 꺼내고, 갓 수정한 다른 알의 아직 합체되지 않은 알과 정자의 핵을 꺼내어 이 핵을 빼낸 수정란에, 먼저 채취한 핵을 이식해서 4일간 배양시킨 후, 다른 흰쥐의 자궁에 옮겨서 3마리의 클론 마우스를 만들어 내었다.
분자유전학적 관점에서 Cloning이란 DNA를 조작하여 조작된 DNA를 증식시켜 원하는 DNA를 다량으로 얻어내는 것이다. 조작된 DNA를 실은 vector를 bacteria에 넣으면 조작된 DNA가 bacteria의 핵 내에 삽입되는 과정을 이용하여 박테리아가 증식할 때 조작된 유전자가 함께 복제되도록 하거나, PCR을 이용하여 DNA를 증폭시킨다. Cloning에 과정에 의해 재조합 된 개체의 모임을 clone이라 부른다.
실험 방법
1. 염분 노출
Cinnamon Clownfish를 17.5psu에 24시간 노출시킨다.(대조군은 저염분에 노출하지 않음)

2. 해부

3. RNA 추출하기
① Trizol 300㎕씩 1.5㎖ tube에 넣는다.
② 조직 0.03g을 핀셋을 이용하여 trizol이 담긴 tube에 넣는다.
③ 조직 homogenize 하기.
④ 실온에서 5분 방치
⑤ Chloroform 60㎕
⑥ Vortex
⑦ 실온에서 3분 방치
⑧ 원심분리 20분
⑨ 위 부분의 맑은 상층액을 튜브로 옮긴다. (150㎕) ☞ RNA층
⑩ Isopropyl alcohol 150㎕
⑪ 실온 10분 방치 후 원심분리 20분
⑫ 밑부분의 흰덩어리(RNA)를 제외한 나머지 액은 버린다.
⑬ EtOH(70%) 300㎕
⑭ 원심분리 5분
⑮ RNA를 제외한 나머지 액은 버린다. : 원심분리기와 비슷한 스핀다운 기계를 이용하여 벽면에 묻어 있는 걸 밑으로 가도록 해서 최대한 뽑을 수 있는 만큼 뽑는다.
⑯ Spin Down을 한다.
⑰ DEPC 30㎕ 넣고 Vortex : 추출된 RNA의 양만큼 넣어준다. 응축된 걸 풀어주고 RNase 저해제 단백질의 화학수식제로서 사용되는 시약이지만 발암성이 있으므로 주의한다.
⑱ -80℃에서 보관한다.
4. RNA 정제단계 & PCR
1) RNA 농도 측정
① Normalize - 2.5 ㎍/㎕. biophotometer를 이용하여 RNA의 값을 구한다
② 첫 번째 튜브 : 0.4518μg/㎕, 두 번째 튜브 : 0.3768μg/㎕
2) cDNA 합성
① DEPC 10㎕, RNA 1㎕ (2.5μg/㎕), Oligo dT 1㎕
② 70℃ 10분 : 반응할 시간을 주기 위함
③ on ice 1분 : 반응을 중지시키기 위함
④ 10x PCR buffer 2㎕, 10mM dNTP 1㎕, 0.1M DTT 2㎕, M-㎖V 0.5㎕
⑤ 42℃ 1시간 : cDNA 합성
⑥ 70℃ 15분
⑦ on ice 1분
⑧ 37℃ 20분
⑨ 4℃ 냉장고 보관
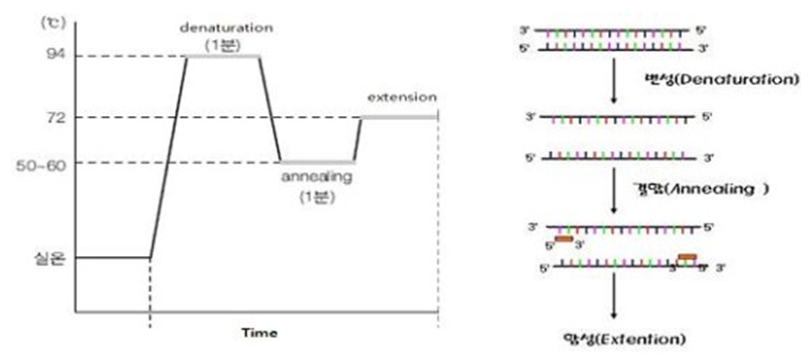
3) PCR (Polymerase chain reaction) - 유전자 증폭 단계
① 94℃-3분, 94℃-30초, 52℃-30초, 72℃-1분(40 cycle), 72℃-7분
② 1 cycle 마다 2배씩 증폭시켜준다.
5. 전기영동 & 겔 정제
1) 전기영동
① 1% 겔만들기
② PCR product와 loading buffer를 섞어서 겔홈에 넣는다.
③ 겔 양 끝에 marker를 넣는다 : 시료의 정확한 분자량을 알아내기 위해 쓰는 이미 분자량을 알고 있는 DNA
④ 약 50분 동안 영동

⑤ EtBr : Gel 생성시 DNA 사이에 들어가서 주황색으로 빛나면서 형광물질 역할
2) Gel Purification : 전기영동을 통해 원하는 DNA를 얻었지만 겔과 함께 섞여있는 상태이므로 DNA를 정제해줘야 한다.
① Gel 무게 3배만큼 GB buffer : gel의 무게 0.03g(gel튜브무게 - 빈 튜브의 무게)
② 50℃에서 약 10분정도 완전히 녹임(중간 중간 vortex)
③ Gel 무게만큼 iso propanol : DNA와 gel의 분리
④ Vortex
⑤ Spin coulumn에 옮기기 : filter 역할
⑥ 실온에서 centrifuge 1분
⑦ 맨 밑에 액 버림
⑧ PW buffer 750㎕ : 거의 알코올이며 세척하는 기능을 한다.
⑨ 실온 5분 방치
⑩ 실온에서 centrifuge 1분
⑪ 맨 밑에 액 버리고 빈 튜브 다시 한번 실온에서 centrifuge 1분
⑫ Spin column을 새 1.5㎖ 튜브에 넣기
⑬ EB buffer 15㎕
⑭ 실온에서 1분 방치 후 centrifuge 1분
⑮ Spin column 버림
6. Ligation
1) Transformation
① 대장균 80㎕에 ligation한 DNA를 모두 넣는다
② 얼음에서 30분간 방치 : DNA가 들어 갈 수 있도록 반응시간을 준다.
③ 42℃에서 45초 heat shock : 대장균이 잠깐 갈라질 때 DNA가 들어간다.
③ 배지에 도말
④ overnight
⑤ 다음날 아침에 균(Colony) 확인

7. Pick up
① LB배지 3㎖씩 분주
② Ampicillin 3㎕씩 분주
③ 이쑤시개로 colony를 찍은 후 새 배지에 살짝 찍는다
④ 튜브에 이쑤시개를 살짝 넣고 흔든다
⑤ 튜브를 37℃로 맞춘 Shaking incubator에 넣는다
⑥ Overnight (16시간)
8. 집균
1) 실험 과정
① 배양된 균을 1.5㎖ 튜브에 적당량 담는다 → 위: 액 아래: 균이 모아진다.
② 실온에서 1분간 Centrifuge
③ 상층액을 버린 후 남은 배양된 균을 1.5㎖ 튜브에 마저 담는다
④ 실온에서 1분간 Centrifuge
⑤ 상층액을 버린 후 냉장 보관된 S1을 250㎕
⑥ Vortex : 희석될 때까지 덩어리를 쪼개기.
⑦ S2 250㎕ 파란색으로 변한다.
⑧ Inverting
⑨ S3 350㎕ 다시 탁한 흰색으로 변한다. (세포 찌꺼기, 대장균 사체)
⑩ Inverting한 후 15분간 Centrifuge
⑪ 상층액을 Spin column에 옮기기
⑫ 실온에서 1분간 Centrifuge
⑬ 하층액 버리기
⑭ PW 750㎕ : 에탄올이 함유되어 있어서 세척되는 과정
⑮ 실온에서 1분간 Centrifuge한 후 하층액 버리고 한번 더 Centrifuge
⑯ 맨위의 튜브를 1.5㎖ 새튜브에 옮긴 후 EB 30㎕
⑰ 1분간 실온 방치 후 실온에서 1분간 Centrifuge → 맨 위의 튜브 버리기
2) 효소처리
① SDW 7㎕, EcoRI buffer 1㎕, EcoRI 0.5㎕, Plasmid DNA 1.5㎕
② Total 10㎕ 37℃ incubator에서 2시간 동안 효소처리 반응 후 전기영동 확인
[분자생물학실험]Gene Cloning 레포트
1. 실험 목적 가. Gene cloning으로 DNA를 자르고 붙이고 변형시켜서 실험에 필요한 재조합 DNA를 제조하고, 기본적인 효소의 처리와 DNA의 분리 및 조작방법 이용하여 분자생물학의 기본을 배우기 위함이다. 2. 실험 이론 및 원리 가. Gene cloning 동식물의 한 개체에서 수정을 거치지 않고, 무성생식에 의하여 양친과 똑같은 유전자 조성을 가진 개체를 얻는 기술이다. 그 개체군을 클론이라고 하며, 꺾꽃이, 접붙이기, 포기나누기 등으로
www.happycampus.com

댓글