Ⅰ. 신약 후보물질의 생산과 품질관리
신약의 품목허가를 받으려면 CTD (Common Technical Document) 에 의거하여 제출 자료를 작성하고 식품의약품안전처장의 허가를 받아야한다. 제출자료의 구성을 나타내는 CTD 체계는 아래와 같으며, 품질에 관한 자료는 제 3부에 있다.
임상시험용 원료와 완제의약품은 임상시험대상자의 안전을 보장하기 위하여, GMP 규정에 의거하여 정확하고 안전한 약품의 공급이 이루어져야 한다. 다만 전임상시험을 위한 원료는 사람에게 투여되는 것이 아니기 때문에 실험실을 포함한 비GMP 시설에서도 생산이 가능하다.

제 3부는 다음과 같이 구성된다.
3.1. 자료목차
3.2. 품질평가보고서
3.2.S 원료의약품에 관한 자료
3.2.S.1 일반정보
3.2.S.2.제조
3.2.S.3. 특성
3.2.S.4. 원료의약품의 관리
3.2.S.4.1 기준
3.2.S.4.2 시험방법
3.2.S.5. 표준품 또는 표준물질
3.2.S.6. 용기 및 포장
3.2.S.7. 안정성
3.2.P. 완제의약품에 관한 자료
3.2.P.1 완제의약품의 개요와 조성
3.2.P.2 개발경위
3.2.P.3 제조
3.2.P.4 첨가제의 관리
3.2.P.5 완제의약품의 품질관리
3.2.P.6 표준품 또는 표준물질
3.2.P.7 용기 및 포장
3.2.P.8 안정성
II. 임상시험용 완제의약품의 생산에 필요한 CMC (Chemical manufacturing Control)
임상시험을 수행하기 위해서는 시험의 단계별로 요구되는 품질관리 정보가 있다. 임상시험의 모든 단계에서 임상시험계획을 검토하는 기본 목적은 임상시험대상자의 안전과 권리를 보장하는 것이며, 이에 따른 정확하고 안전한 약품의 공급이 이루어져야 한다. 「의약품 등의 안전에 관한 규칙」제24조제1항제2호에 따른 별표 1의 의약품 제조 및 품질관리기준에 맞게 제조되었음을 증명하는 서류 또는 자료가 요구 된다.
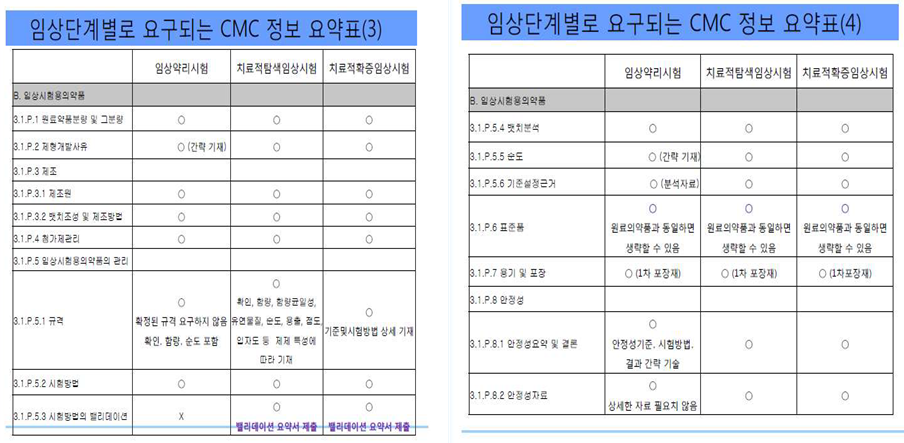
임상시험용 완제의약품의 생산과 품질관리에 관한 자료는 다음의 내용을 포함한다.
1) 임상시험용의약품의 개요와 조성(Description and Composition of the Investigational Medicinal Product) : 임상시험용 의약품의 정성적 및 정량적 조성을 서술한다. 제형 (dosage form)에 대한 간략한 서술 또는 표, 그리고 각 첨가제의 기능이 포함되어야 한다.
2) 개발 경위(Pharmaceutical Development) : 새로운 제형(new pharmaceutical form) 또는 첨가제를 사용한 근거를 포함하여 제형(formulation) 개발 경위를 간략하게 서술한다. 개발 초기에는 이 장에 포함될 정보가 없거나 제한적일 수 있다.
3) 제조공정 개발(Manufacturing Process Development) : 제1상 및 제2상 임상시험에서 각각 사용한 제조 공정과 비교할 때 현 제조 공정이 변경되었을 경우 이를 설명한다. 잠재적인 임상 관련성이 있는 제형(dosage form) 특이적인 품질 파라미터 (예: in vitro 용출률)가 변경된 경우에는 특별한 고려를 한다.
4) 제조원(Manufacturer(s)) : 수탁업소와 각 사업소를 포함하여 제조 및 시험에 관여하는 모든 제조원의 명칭, 주소 및 책임 범위를 기재한다. 복수의 제조원이 임상시험용의약품의 제조에 참여 할 경우, 각각의 책임 부과 범위를 명확하게 서술할 필요가 있다.
5) 배치 조성(Batch Formula) : 임상시험에서 사용하는 제조단위에 대해 배치 조성을 기재한다. 관련성이 있을 경우, 적절한 배치 크기의 범위를 제시할 수 있다.
6) 제조공정 및 공정관리 개요(Description of Manufacturing Process and Process Controls)
각 단계에서 사용하는 성분과 모든 관련 공정관리를 포함하는 공정 흐름도를 기재한다. 이와 더불어, 제조 공정의 간략한 서술적 설명이 포함되어야 한다. 비-표준적 제조 공정 또는 신기술이나 새로운 포장 공정이 사용될 경우 더 상세하게 서술해야 한다.
7) 주요공정 및 반제품 관리(Control of Critical Steps and Intermediates)
이 정보는 제1상 및 제2상 임상시험에서는 아래의 경우를 제외하고 요구되지 않는다.
• 비-표준적 제조 공정
• 무균 제제의 제조 공정
8) 공정밸리데이션 및 평가(Process Validation and/or Evaluation) : 이 자료는 식약처장이 인정하는 공정서에 서술되지 않은 비-표준적 멸균 공정과 비-표준적 제조 공정을 제외하고는 개발 단계(제1상, 제2상, 제3상 임상시험 단계)에서 요구되지 않는다. 이에 해당되는 경우, 주요 제조 단계, 제조 공정의 밸리데이션, 그리고 적용하고 있는 공정 관리를 서술한다.
9) 첨가제의 관리(Control of Excipients)
10) 기준(Specification) : 식약처장이 인정하는 공정서 규격을 인용하여 기재할 수 있다. 식약처장이 인정하는 공정서 규격의 물질로 구성된 첨가제 혼합물 (예: 필름-코팅용 예비-제조 건식 혼합물)은 혼합물의 일반 기준으로 충분할 것이다. 앞서 언급한 규격에 해당되지 않는 첨가제는 자사 기준(in-house monograph)을 기재한다.
11) 시험방법(Analytical Procedures) : 약전의 일반시험법을 인용할 수 없는 경우, 사용한 시험방법을 기재한다.
12) 새로운 첨가제(Novel Excipients) : 새로운 첨가제는 완제의약품의 안전성과 관련이 있는 제조 공정, 특성 및 관리에 대해 상세하게 기재한다.
13) 임상시험용 의약품의 관리(Control of Investigational Medicinal Product)
14) 기준(Specifications)
선택된 출하 및 사용기간 기준(shelf-life specifications)을 제출한다 (시험법 및 허용 기준 포함). 개별 분해 산물과 분해 산물의 합에 대한 상한치를 설정할 수 있다. 안전성 고려사항을 검토해야 하며, 허용치(limits)는 비-임상/임상시험에 사용된 원료의약품 배치의 불순물 프로파일에 근거한다. 기준(specifications) 및 허용 기준(acceptance criteria)은 이후 개발 과정동안 검토하고 조정되어야 한다.
III. 임상시험용 원료의약품의 생산과 품질관리
전술한 임상시험용 완제의약품의 생산을 위한 원료의약품의 품질관리에 관한 자료는 다음의 내용을 포함한다.

일반 정보(General Information)
1) 명칭(Nomenclature) : 원료의약품의 명칭에 관한 정보를 기재해야 한다.(예: INN-명, 약전 수재명, 화학명 [IUPAC, CAS-RN], 실험실 코드, 이명 또는 코드)
2) 구조(Structure) : 각각의 개발 단계에서 수집된 자료를 기재한다. 구조식, 분자량, 규명이 가능한 키랄성/입체화학을 포함하여 기재한다.
3) 일반적 특성(General Properties) : 원료의약품의 물리-화학적 및 기타 관련 특성, 특히 용해도, pKa, 결정 다형, 이성질체, log P, 투과도 등과 같이 약리학적 또는 독성학적 안전성에 영향을 줄 수 있는 물리-화학적 특성에 대한 목록을 기재한다.
4) 제조원(Manufacturer(s)) : 수탁업소와 각 사업소를 포함하여 제조 및 시험에 관여하는 모든 제조원의 명칭, 주소 및 책임 범위를 기재한다.
5) 제조공정 및 공정관리(Description of Manufacturing Process and Process Controls)
화학 물질은 합성 공정의 간략한 요약과 각 단계에서 사용하는 출발 물질, 중간체, 용매, 촉매 및 주요 시약이 포함되어 있는 공정 흐름도를 기재한다. 관련 공정관리도 모두 기재한다. 합성의 주요 단계가 확인되었을 경우 더 상세하게 기술되는 것이 적절하다. 해당되는 경우 출발물질의 입체-화학적 특성이 포함되어야 한다. 식약처장이 인정하는 공정서의 규격에 적합한 물질은 추가 요구 사항이 없다. 임상시험에서 사용되는 배치(batch) 크기의 범위나 생산 규모를 서술한다.
6) 원료물질 관리(Control of Materials)
원료의약품의 제조에 사용하는 물질 (예: 원료물질, 출발물질, 용매, 시약, 촉매)의 목록을 작성하고 중요하다고 예측되는 모든 속성(attributes)의 품질 및 관리에 대해 간략하게 요약한다. (예를 들어, 원료의약품 중 불순물을 제한하기 위해 관리가 필요한 경우 [예: 키랄성 관리, 금속 촉매 관리 또는 전구물질의 관리부터 잠재적인 유전독성 불순물까지])
7) 주요공정 및 중간체 관리(Control of Critical Steps and Intermediates) : 합성의 주요 단계별로 이를 관리하기 위한 시험 및 허용 기준을 간략하게 요약한다.
8) 제조공정 개발(Manufacturing Process Development)
제조공정이 비임상시험시 사용된 배치의 제조공정과 차이가 있을 경우 이를 문서화한다. 이 경우 비임상시험에 사용된 원료의약품의 제조 공정 흐름도를 기재한다.
9) 구조 및 기타 특성(Elucidation of Structure and other Characteristics) : 화학적으로 정의된 물질의 구조는 적합한 방법론으로 확립되어야 한다; 관련 자료가 제공되어야 한다.
10) 순도(Impurities) : 식약처장이 인정하는 공정서 규격에 적합한 물질은 추가 요구되는 사항이 없다. 위에 언급한 약전에 수재되지 않아 인용할 수 없는 경우, 임상시험에서 사용하는 원료의약품과 관련된 출발물질 또는 제조공정으로부터 유래하는 불순물, 분해 산물 및 잔류 용매를 서술한다.
11) 기준(Specification) : 임상시험에서 사용하는 원료의약품 배치의 기준, 사용하는 시험 및 허용 기준을 명시한다. 확인 및 함량 시험은 필수이다. 불순물은 안전성을 고려하여 상한치를 설정한다. 이런 기준들은 이후 개발 과정에서 재검토하고 조정할 필요가 있을 수 있다. 무균 제제의 제조에 사용하는 원료의약품은 미생물학적 품질을 명시한다.
12) 시험방법(Analytical Procedures) : 원료의약품 기준에 포함되어 있는 모든 시험에 대해 사용하는 시험법을 서술해야 한다 (예: 역상-HPLC, 전위차 적정, 헤드스페이스-GC 등). 시험방법을 상세하게 기술할 필요는 없다. 식약처장이 인정하는 공정서 규격에 적합한 물질은 해당 규격을 인용하는 것으로 충분할 것이다.
13) 시험방법 밸리데이션(Validation of Analytical Procedures) : 제1상 임상시험의 경우, 사용된 시험법의 적합성(Suitability)이 확인되어야 한다. 시험방법 밸리데이션을 수행하기 위한 허용 한도(예: 관련성이 있는 불순물 함량 측정에 대한 허용 한도) 및 파라미터(적절할 경우, 특이성, 직선성, 범위, 정확성, 정밀성, 정량한계 및 검출한계)를 표 형식으로 기재한다.
14) 배치 분석(Batch Analyses) : 현 임상시험 및 비임상시험에 사용하는 배치, 그리고 해당될 경우 선행 임상시험에서 사용한 모든 배치에 대한 시험성적서 또는 배치 결과를 제시한다. 현 임상시험에서 사용하는 배치에 관한 분석자료가 없는 경우 대표 배치의 자료를 제출할 수 도 있다. 배치 번호, 배치 크기, 제조소, 제조 일자, 관리 방법, 허용 기준 및 시험 결과를 목록으로 작성한다. 각 배치에 사용한 제조 공정을 기재한다.
15) 기준 설정근거(Justification of Specification(s)) : 약전을 인용할 수 없는 물질은 완제의약품의 성능과 관련이 있을 수 있는 불순물 및 모든 기타 파라미터에 대해 불순물의 관리에 사용한 방법뿐만 아니라 안전성 및 독성 자료에 근거하여 간략한 기준 설정 근거 및 허용 기준을 기재한다. 합성에 사용한 용매 및 촉매도 고려한다.
16) 표준품 또는 표준 물질(Reference Standards or Materials) : 해당될 경우, 표준품을 확립하기 위해 사용한 원료의약품 배치의 특성 파라미터를 기재한다.
17) 용기 및 포장(Container Closure System) : 원료의약품에 사용하는 일차 포장재를 서술한다.
18) 안정성(Stability) : 각각의 개발 단계에서 입수된 안정성 자료를 표로 요약한다. 원료의약품의 안정성에 중요하다고 알려진 파라미터, 즉 화학적 및 물리적 민감도 (예: 광과민성, 흡습성)를 제시할 필요가 있다. 잠재적인 분해 경로를 서술한다. 그렇지 않더라도 원료 의약품이 약전 규격에 해당될 경우, 사용시점에서 원료의약품이 기준을 충족한다는 확증이 있으면 받아들여진다.
IV. 전임상 원료의약품의 생산
신약 후보물질이 발굴되어 전임상시험을 수행하게 되면 시험에 필요한 대량의 원료를 확보하기 위한 작업이 필요하다. 초기 후보물질의 발굴에 적용되었던 의약화학적인 방법은 필요에 따라 공정개선이 이루어져야 한다. 원료의 소요량은 전임상시험의 내용과 투여 용량에 의존적이나, 기본적으로 독성시험과 pre-formulation 연구에 수 백그램의 원료가 필요하다. 따라서 대용량의 합성에 알맞은 반응시약, 용매 및 정제공정의 개선이 요구된다.
대량의 화합물을 정제하기에 보다 유용한 재결정 등의 방법이 정제에 요구된다. 품질관리를 위한 기준 및 시험법의 작성을 위한 순도, 역가시험 등이 필요하다. 순도 관리는 보통 95% 이상을 요구하나 따로 정해진 범위는 없다. 다만 임상시험을 위한 순도가 더 높아야 하므로 전임상단계에서 지나치게 높은 순도를 설정하지는 않는다. 또한 역가를 확보하기 위한 표준물질의 합성이 요구된다. 전임상단계에서의 생산은 비GMP조건에서 이루어져도 무방하며, 공식적인 제조기록서는 요구되지 않으나, 제조공정을 상세하고 정확하게 기록하여야 한다.
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 신약개발의 프로세스
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 신약 후보물질의 발굴
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 완제 및 원료의약품 GMP
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 비임상 효력시험
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 일반약리시험과 안전성약리시험
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 흡수분포대사배설시험 및 약물상호작용시험
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 독성시험 (비임상 안전성평가시험)
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 비임상시험관리기준 (GLP)
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 비임상시험과 임상시험의 연계
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 임상개발 개요
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 임상시험의 과학적 측면
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 개별 Product level

댓글