임상개발은 시야 , 즉 scope 에 따라 개별적인 임상시험 , 하나의 의약품 을 개발하기 위해 여러 개의 임상시험을 목적 , 단계별로 시행하는 제품 수준의 임상개발 , 여러 임상개발 프로젝트를 통합적으로 관리하는 포트폴리오 수준으로 나눌 수 있습니다. 본 내용은 임상개발의 기본 요소인 임상시험부터, 제품 수준의 임상개발, 임상개발 프로젝트의 포트폴리오 구성 순으로 진행될 예정입니다
미국 국립보건원의 정의에 따르면 임상시험이란 임상연구보다는 더 좁은 의미로 쓰이며, 인체에 적용되는 예방, 진단, 치료를 위한 물질, 기구, 또는 방법의 주된 효과와 가치를 평가하기 위한 과학적인 연구활동입니다. 디벨롭의 의미는 제거한다는 의미의 디 (de)와 싸다라는 의미의 벨롭 (velop)이 결합된 단어로서, 말 그대로 싸여 있는 것을 풀다, 밝히다는 의미입니다.
따라서 임상개발 즉 clinical development는 의약품, 의료기기, 진단 수단 개발의 한 과정으로서 임상시험을 기본 단위로 하여, 여러 임상시험을 단계별로 시행하여 이들을 조합하여 후보물질이 가지는 임상적 특성을 밝히는 즉 규명해 나가는 절차 또는 과정으로서, 단어의 의미 그대로 임상개발은 discovery과정에서 도출된 후보물질의 임상적 특성을 밝히는 과정으로 정의될 수 있습니다. 임상개발은 복합적이고 장기간이 소요되는 과정으로 구체적인 목표설정과 효율적인 진행, 적시의 판단이 필요한 과정입니다. 더욱이 시험대상자 역시 인간이라는 점에서 윤리적인 측면도 중요한 고려 사항이 됩니다.
개별 Trial level
임상 시험은 계획한 목적을 달성하기 위해 헬싱키선언과 임상시험관리기준에서 언급된 윤리적 원칙과 과학적 원칙에 따라 설계 실시 및 분석되어야 합니다. 따라서 본 내용에서는 윤리적 측면과 임상시험 설계 시 고려되는 과학적 측면으로 나누어 임상시험에 대해 소개하고자 합니다
1. 임상시험의 윤리적 측면
본 내용에서는 우선 임상시험 윤리가 정립되어 온 역사적 배경에 대해 설명하고 임상시험관리기준, 영어로 는 good clinical practice 약어로 는 GCP 에 대해 소개하고자 합니다
1) 임상시험 윤리와 관련된 역사적 배경
2차 세계대전 중 나치 통제하에서 유태인을 포함한 수용소 수감자에게 비윤리적인 절차로 연구를 수행한 지휘관, 의사 및 보건행정 공무원을 대상으로 1946년 12월 독일 뉘른베르크에서 열린 전범재판과정 중 판결문을 통해 ‘허용 가능한 의학실험’에 대한 기준이 제시되었습니다. 허용 가능한 의학실험은 뉘른베르크 강령이라는 이름 하에 10개 항이 제시되었으며, 주요 내용은 자발적인 참여 및 동의서 요건, 적절한 위험 이익분석이 인체 대상 연구 전 시행되어야 한다는 점, 언제든 조건 없이 연구참여를 그만 둘 수 있는 권리가 있다는 점이 제시되었습니다.
뉘른베르크 강령은 이 후 제네바 선원 및 헬싱키 선언 등 사람을 대상으로 하는 연구의 윤리 기준의 지침을 마련하는 개기가 되었습니다. 1954년 발표된 제네바 선언은 재판과정 중 법률가들에 의해 제시된 연구 윤리 기준이 아니라 임상시험의 실제 연구자인 의사들이 제정한 자율적인 연구 윤리라는 의미가 있습니다. 헬싱키 선언의 주된 골자는 뉘른베르크 강령에 덧붙여 첫째 시험대상자의 이익은 사외의 이익보다도 항상 우선권을 가진다는 것, 둘째 임상연구의 모든 시험대상자는 알려진 최상의 치료를 받아야 한다는 점이 추가되었습니다.
제네바 선언을 바탕으로 1964년 헬싱키 세계의사협회 제 18차 총회에서 임상연구자인 의사들의 자체적이며 국제적인 연구윤리 강령인 헬싱키 선언이 제정되었으며, 헬싱키 선언은 이후 GCP의 가장 기본적인 윤리 원칙으로 자리잡게 되었습니다. 헬싱키 선언은 현재 7차까지 개정되었습니다.
나치의 비윤리적인 인체실험의 반성에서 유래된 뉘른베르크 강령과 제네바선언 및 헬싱키선언이 이미 1946년, 1954년, 1964년 발표된 이후에도 비윤리적 임상시험은 지속되고 있었습니다. 1932년부터 미국 켄터키 주의 터스키키 시에서 시행된 흑인대상의 매독연구는 뉘른베르크 강령, 제네바 선언, 헬싱키 선언 이후에도 이미 페니실린이 매독치료에 널리 사용되고 이었음에도 치료 없이 추적 관찰한 결과가 의학저널에 보고되었으나 윤리적 측면에서 문제 시 되지 않았고, 1972년 미국 공중보건원 직원의 폭로에 의해서 문제가 제기되었습니다.
터스키키 매독연구에 대한 폭로 이후 1974년에 미국에서는 National Research Act가 통과되었으며, 생명의학 및 행동 연구 관련 인간 보호를 위한 국가위원회가 발족되고 이 위원회는 1979년 벨몬트 리포트를 내놓게 됩니다. 벨몬트 리포트는 인간을 포함하는 연구와 관련된 기본 윤리 원칙, 연구와 실제 임상간의 경계, 위험/이득에 대한 기준, 동의서에 대해 지켜야 할 윤리 원칙과 수행에 대해 다루고 있습니다. 이후 1981년 미국 연방규정집 Code of Federal Regulations에 인간피험자 보호를 위한 일반규정이 별도로 제정되었습니다.
2) Good Clinical Practice(GCP) 정의
헬싱키 선언을 비롯하여 여러 강령 등을 통해 정립된 임상시험의 윤리적 측면과 미국 FDA의 제약회사 실사를 통해 발견된 과학적 신뢰도 측면에서의 여러 문제점이 발견된 것을 계기로 Good clinical practice 약자로 GCP가 제정되게 됩니다.
GCP란 인간이 시험 대상으로 참여하는 임상시험의 설계, 진행, 기록 및 보고에 대하여 규정하고 있는 윤리 및 과학적 질에 관한 국제적 표준을 의미하며, GCP를 준수한다는 것은 헬싱키 선언에 기원한 원칙에 부합되도록 시험대상자의 권리와 안전 및 안녕이 보호되었고 임상시험 데이터가 신뢰할 수 있음을 공식적으로 보장한다는 것을 의미합니다.
3) 국내 GCP 소개
국내 GCP는 의약품임상시험관리기준을 통해 제시되었습니다, 의약품임상시험관리기준은 1987년 공포되었으나 국내 여건 상 시행이 보류되다가 1995년 전면 시행되었습니다. 현재는 의약품 등의 안전에 관한 규칙 (총리령 제1022호) 중 별표 4에 해당하는 내용입니다. 국내 GCP인 의약품임상시험관리기준과 국제적 표준인 ICH의 GCP는 규정 에 언급되어 있는 사안에 대해서는 유사한 내용을 담고 있으나, 일부 사안에 대한 내용이 포함 또는 미포함 되어 있다는 점에서 차이가 있습니다. 예컨대, 국내 규정에는 ‘임상시험의 계약 및 임상시험 실시기관’이 별도 항목으로 제시되어 있으나, ICH GCP에 포함되어 있는 임상시험 계획서와 시험자자료집 관련 내용은 포함되어 있지 않습니다.

4) GCP 원칙
ICH와 국내 규정간의 임상시험에 대한 GCP 원칙은 매우 유사합니다. GCP 원칙은 임상시험 수행 도중 지켜져야 하며 그 내용은 다음과 같습니다.
① 임상시험은 헬싱키 선언에 근거한 윤리규정, 임상시험관리기준 및 관련규정에 따라 수행되어야 한다.
② 임상시험으로부터 예측되는 위험과 불편 사항에 대한 충분한 고려를 통해 피험자 개인과 사회가 얻을 수 있는 이익이 그 위험성을 상회 또는 정당화할 수 있다고 판단되는 경우에 한하여 임상시험을 실시하여야 한다.
③ 피험자의 권리․안전․복지는 우선 검토의 대상으로 과학과 사회의 이익보다 중요하다.
④ 해당 임상시험용의약품에 대한 임상 및 비임상 관련 정보는 실시하고자 하는 임상시험에 적합한 것이어야 한다.
⑤ 임상시험은 과학적으로 타당하여야 하며, 임상시험계획서는 명확하고 상세히 기술되어야 한다.
⑥ 임상시험은 식품의약품안전청장 (ICH: IRB, IEC이 승인한 임상시험계획서에 따라 실시하여야 한다.
⑦ 피험자에게 제공되는 의학적 처치나 결정은 의사 등 (ICH: 자격을 갖춘 의사, 경우에 따라 치과의사) 의 책임하에 이루어져야 한다.
⑧ 임상시험 수행에 참여하는 모든 사람들은 각자의 업무 수행을 위한 적절한 교육․훈련을 받고, 경험을 갖고 있어야 한다.
⑨ 임상시험 참여 전에 모든 피험자로부터 자발적인 임상시험 참가 동의를 받아야 한다.
⑩ 모든 임상시험 관련 정보는 정확한 보고, 해석, 확인이 가능하도록 기록․처리․보존되어야 한다.
⑪ 피험자의 신원에 관한 모든 기록은 비밀보장이 되도록 관련규정에 따라 취급하여야 한다.
⑫ 임상시험용의약품은 의약품제조 및 품질관리기준(GMP)에 따라 관리되어야 하며, 식품의약품안전청장이 승인한 임상시험계획서 (ICH: 승인 받은 계획서)에 따라 사용되어야 한다.
⑬ 임상시험은 신뢰성을 보증할 수 있는 체계하에서 실시되어야 한다 입니다.
5) GCP 위반 : 미국 FDA 실사 결과
미국 FDA가 시행한 실사 결과 지역마다 차이가 있으나 가장 흔하게 발견되는 GCP 위반사항은 임상시험 관련 기록의 미비 (inadequate and incorrect records), 계획서 미준수 (failure to adhere to protocol), 이상반응 보고 절차 위반 (failure to report adverse reactions)이었습니다.
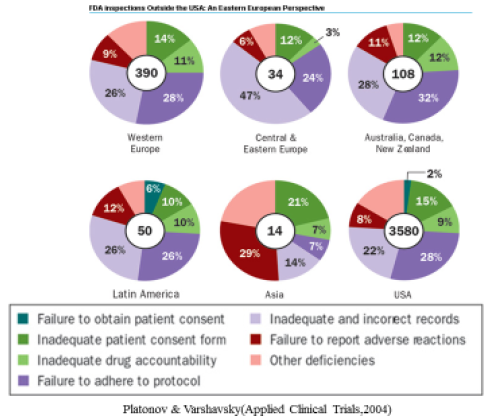
슬라이드로 별도 제시하지는 않았으나 김영옥 등이 2005년도에 발표한 바에 따르면 국내 GCP 위반사항의 경우 시험자 관련 사항이 60%, IRB 관련사항과 의뢰자 관련 사항이 각각 약 20%정도였으며, 시험자 관련하여서는 임상시험 관련 기록의 미비가 가장 빈번하였고, IRB는 SOP 관련사항, 의뢰자는 임상시험용 의약품과 관련된 사항이 가장 빈번하였다. 현재는 GCP 준수와 관련하여 국내 상황이 많이 개선된 것으로 판단됩니다.
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 신약개발의 프로세스
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 신약 후보물질의 발굴
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 신약 후보물질의 생산과 품질관리
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 완제 및 원료의약품 GMP
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 비임상 효력시험
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 일반약리시험과 안전성약리시험
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 흡수분포대사배설시험 및 약물상호작용시험
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 독성시험 (비임상 안전성평가시험)
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 비임상시험관리기준 (GLP)
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 비임상시험과 임상시험의 연계
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 임상시험의 과학적 측면
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 개별 Product level

댓글