1. 임상개발의 단계와 이와 관련된 임상시험의 종류를 이해한다.
2. 시험약을 최초로 인체에 적용하는 임상시험에서의 주요 결정사항에 대해 이해한다.
3. 1c 임상시험의 목적에 대해 이해한다.
4. 생물약제학 임상시험의 목적에 대해 이해한다.
5. adequate and well controlled 임상시험의 구성요소와 의미를 이해한다.
6. 임상시험 데이터의 종류를 이해하고 중요함을 인식한다.
임상개발의 단계
하나의 약물을 개발하는 과정의 대략적인 과정은 그림과 같습니다.
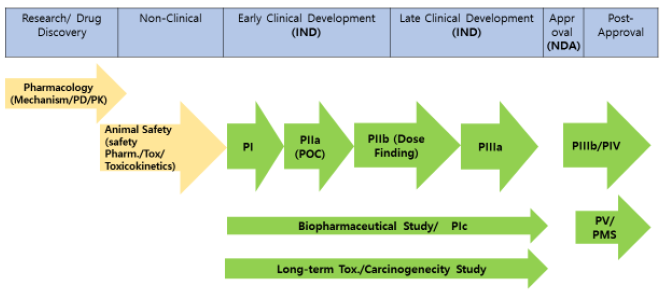
요약하자면, discovery 후, 비임상 독성시험을 수행하고 안전성을 평가한 다음 임상개발 과정에 진입하게 됩니다. 각 단계 임상시험을 진행하는데 필요한 전임상시험 자료의 범위는 허가 기관의 가이드라인 등을 통해 제시되어 있습니다. 임상개발은 1상, 2상, 3상의 단계를 거쳐 품목승인을 신청하게 되며, 품목승인 후에는 lifecycle management 차원에서 후기 3상 (IIIb)이나 허가 후 임상시험 (IV)을 수행할 수 있으며, 시판후 약물의 안전성을 평가하기 위한 약물안전성관리 (pharmacovigilance) 또는 PMS (postmarketing surveillance) 업무를 진행하여야 합니다. 임상시험의 종류는 전술한 바와 같이 시험이 실시되는 단계에 따라 분류할 수도 있고, 목적에 따라 분류할 수도 있습니다.
임상시험의 목적에 따른 임상시험의 종류

임상약리시험은 1상 임상시험과 생물약제학 임상시험을 포함합니다.
1상 임상시험은 단회 투여 임상시험인 1a 임상시험, 반복투여 임상시험인 1b 임상시험, 그리고 기타 약물대사와 상호작용, 특수인구집단에서의 약동, 약력학 정보를 획득하기 위한 1c 임상시험 등을 포함합니다. 초기임상시험인 Ia 임상시험과 Ib 임상시험은 기본적으로 인체에서의 Tolerability를 측정하기 위한 시험이며, 궁극적으로 여러 1상 결과를 종합 평가하여 2상에서 적용할 RP2D (recommended phase 2 dose) 결정하기 위한 것입니다.
또한 약력학적, 약동학적 특성을 연구하고, 가능하면 dose-limiting adverse events의 profile을 제한적으로나마 알고자 하는 것이 목적입니다. 그러나 피험자의 수가 적으므로 full adverse events profile을 알기는 어려움이 있습니다. 2상 임상시험은 치료적 탐색 임상시험에 해당합니다. 2상 임상시험은 2a 임상시험과 2b 임상시험으로 분류할 수 있습니다. 2a 임상시험은 환자에서 proof of concept을 하기 위한 임상시험으로 환자에서 치료제로서 가능성을 탐색하기 위한 임상시험이며 POC 임상시험이라고 불리며, 2b 임상시험은 3상에서 사용된 용량을 결정하기 위한 임상시험으로 dose finding 임상시험으로도 불린다.
3상 임상시험은 치료적 확증 임상시험에 해당하며, 최초 폼목 승인을 위한 3a 임상시험과 품목승인 후 적응증을 추가하기 위한 3b 임상시험을 포함합니다. 4상 임상시험으로 치료적 사용 임상시험이 있습니다. 치료적 사용 임상시험은 유효성 즉 efficacy아닌 유용성 effectiveness에 대해 연구하거나 약물경제학 및 기타 의약품의 사용과 관련된 부가적인 평가항목에 대한 임상시험을 의미합니다. 초기 임상시험의 부가적인 설명과,Ic 임상시험과 생물약제학 임상시험 기타 임상시험은 다음 슬라이드에서 설명하고자 합니다.
인체 최초 투여 임상시험
인체 최초 투여 임상시험시 주요한 결정사항은 시험대상자의 선정, 초회투여용량, 용량증량방법, 투여경로, 투여기간, 관찰 사항 등입니다.

초회투여용량과 관련하여 건강한 시험대상자를 대상으로 인체에 최초로 약물을 투여할 경우 첫 투여 용량은 일반적으로 NOAEL을 기반으로 결정합니다.
NOAEL (노엘로 발음) 또는 무독성량은 No Observed Adverse Effect Level 의 약자로서 일반적으로 대조군에 비해 이상반응이 유의하지 않게 증가하는 가장 높은 용량 수준을 의미합니다. NOAEL을 기반으로 인체 최초투여용량을 설정하는 절차는 독성실험에서 각 동물 종의 NOAEL 결정, 각 동물에서의 NOAEL에 대한 Human equivalence dose (HED) 계산, 이중 가장 적당한 동물 종 선정, safety factor의 적용, MRSD 산정, PAD를 고려한 clinical starting dose 산정의 순으로 진행합니다.
비임상 독성시험에서 NOAEL 설정에 사용된 부작용은 건강한 성인 자원자에게 초회 투여 시 관찰되어서는 안 되는 작용을 대상으로 설정합니다. 시험대상자 선정과 관련하여 시험용 의약품을 최초로 인체에 적용할 때 시험대상자는 일반적으로 환자보다는 건강한 자원자를 우선적으로 고려합니다. 건강한 자원자의 경우 환자에 비해 독성에 대한 저향력이 강하므로 이상반응에서의 회복력이 더 양호할 것으로 판단되며, 시험용 의약품에 대한 연구를 위해 필요한 많은 채혈에 대해 보다 내성이 높을 것으로 판단되기 때문입니다. 또한 환자를 대상으로 시험대상자를 모집하는 것보다 더 속도가 빠른 것도 장점입니다.
또한 노령층이 occult disease에 이환되어 있을 가능성이 더 높고 신장, 폐 기능 저항로 인한 약물 제거 능력이 감소되어 있다는 점을 고려하여 노인보다는 젊은 시험대상자가 선호됩니다. 용량증량방식과 관련하여 1,2,3,5,8, 13, 21배식으로 용량이 증량되는 Fibonacci Method, 2배, 1.67배, 1.50배. 1.33배식으로 용량이 증량되는 Modified Fibonacci Method, 두배씩 증량된 Doubling Dose Escalation, 약 1배, 3배, 10배, 30배,…식으로 용량이 증량되는 Semi-Log “Fast-track” Dose Escalation 방식 등이 있습니다.
투여경로와 관련하여 동물시험에서 사람 투여경로와 동일 경로 시험 시행되어야 하며, 임상약리 시험 역시 추후 예상 투여 경로 투여로 시행되어야 합니다. 단 투여 경로와 관련하여 IV INFUSION이 선택 될 때 얻을 수 있는 부수적인 장점으로 인하여 추호 예상 투여 경로와 상관 없이 종종 이용되기도 합니다. 투여기간은 일반적으로 비임상 독성시험에서 평가된 기간 이내에서 투여하며 이에 대한 가이드라인이 별도로 제시되어 있습니다.
초기 임상시험에서 가장 중점을 두고 관찰하는 사항은 내약성 정보인 MTD (maximum Tolerable Dose)이며 이는 Dose limiting toxicity가 유의하게 증가하는 농도를 의미합니다. DLT (Dose limiting toxicity)는 미리 정하는 경우가 많으며, 예를 들어 NCI common Terminology for Adverse Events상으로 Grade 3에 해당하는 독성으로 정하는 경우가 많다. 제1상 임상시험에도 가능하면 약력학 변수를 측정합니다. 예를 들어, 항고혈압제의 경우에는 혈압, 맥박수, 전신말초혈관저항(total peripheral resistance, 쎾) 등을 비관혈적으로 측정할 수 있고, 궤양치료제의 경우24시간이상 위산도를 측정할 수 있습니다. 기전에 따라 다르기는 하지만, 당뇨병치료제의 경우에는 표준식사 후의 혈당,인슐린 변화에 대해 시간에 따른 추이를 관찰할 수 있습니다. 항생제의 경우에는 건강자원자에서 채혈한 혈액을 임상분리균주에 적용하여 항생효과를 관찰할 수 있습니다.
일반적으로 용량증량 방법 기준 1상 임상시험 Design 분류

1상 임상시험에 용량증량 방법과 관련하여 일반적으로 널리 사용되는 3+3 설계 이외에도 여러 방식들이 사용되고 있습니다. 3+3방식이외에도 simple up and down 방식, accelerated titration 방식, pharmacologically guided dose escalation 방식, modified continual reassessment 방식, 예시된 escalation with overdose control을 포함하는 adaptive 방식 등이 있습니다. 슬라이드에서는 현재 사용되고 있는 용량증량 방식에 대해 개략적으로 표시한 것입니다.
Phase lc 임상시험
약물의 Exposure, Drug Effects에 영향을 미칠 수 있는 인자들과 인자가 미치는 영향을 알아보기 위한 임상약리 시험들

임상개발 도중 시험대상자는 선정기준에 따라 모집하게 됩니다. 결과적으로 임상시험에 포함된 시험대상자의 범위는 제한적이므로 임상시험 대상 적응증의 모든 환자들을 대변하지 못하며 외적타당도가 제한적일 수 밖에 없다. 또한 치료적 확증을 위한 임상시험에서 제외된 인구집단이 시판 후 임상시험에서 평가된 의약품을 시판 후 투여 받을 가능성이 있습니다. 그러나 다양한 환자특성 별로 대규모의 치료적 확증 임상시험을 수행하는 것은 시간이나 경제적 측면에서 어려운 점이 많다.
따라서 임상약리시험인 1c 임상시험을 수행하고 환자 특성이 약물의 exposure에 미치는 영향을 알아보는 약물동태학 임상시험을 실시하고, 약물 exposure와 약물효과간의 관계를 이용하여 결과적으로 환자특성이 약물효과에 미치는 영향을 외삽하여 평가하는 것이 필요하다. 환자의 특성은 크게 내인성과 외인성 인자로 나뉘며, 내인성 인자에는 나이, 인종, 간장해, 신장해, 질병상태, 임신 및 수유 여부, 성별, 유전형 등이 있으며, 외인성 인자에는 약물상호작용, 식사, 알코올, 흡연, 의료환경 등이 있습니다.
예컨데 신장해 환자에서의 약동학 특성 및 제한적이나마 소수에서의 안전성 프로파일 정보를 생성하고, 신장해 정도에 따른 약동학 특성과 장해가 없는 자원자의 약동학을 비교합니다. 이를 통해 신장해 환자에서 약물용량 조절이 필요한지 여부에 대해 알수 있을 것입니다.
생물약제학 임상시험
제형, 투여경로 등이 처음 계획하였던 바대로 항상 유지되는 것은 아니며, 신약을 개발하는 동안 변경되는 경우가 매우 빈번하다. 이 경우 상이한 제형을 통해 획득한 정보가 유의성을 확보하기 위해서는 생물약제학 평가를 통해 상이한 제형 투여에 따른 약동학 차이 발생 여부를 검토하여야 합니다. 생물약제학 범주에는 생물학적 동등성 시험, 생체이용률에 관한 시험, 경구의약품의 경우 비교용출 시험 등이 포함됩니다.
생물약제학 자료는 CTD 상에서 2.5.2와 2.7.1에서 요약되며, 5에서 임상시험보고서가 첨부됩니다. 생물약제학 자료 구성과 관련된 법규 및 가이드라인은 의약품동등성 시험 관리 기준, 생체이용률에 미치는 음식물 영향 및 식후 생물학적동등성 시험 가이드라인, 경구용의약품의 용출규격 설정 가이드라인 등이 있습니다.
Adequate and Well-Controlled Clinical Investigation
adequate and well controlled 임상시험이란, 첫째 명확한 시험목적이 제시되어야 하며, 둘쨰, 대조군과 타당성이 있는 비교를 할 수 있는 적절한 시험 설계가 되어야 하고, 시험대상자 선정이 적절하여야 하며, 비뚤림이 최소화되도록 장치가 마련되어야 하며, 치료에 대한 시험대상자의 반응에 대해 정확히 규정되고 신뢰성 있는 평가법이 동반된 임상시험을 의미합니다.
임상시험의 데이터
임상시험 데이터는 일반적으로 무작위배정 전 수집되는 데이터로 인구학적 정보, 동반질환 정보, 결과에 영향을 줄 수 있는 예후인자에 대한 변수인 기저치 변수 (Baseline Variables), 무작위배정 후 순응도, 교란을 줄 수 있는 인자에 대한 정보, 안전성 모니터링을 위한 정보 등인 공정변수 (Process Variables), 임상시험에서 평가되는 치료에 의해 결정되는 변수들인 결과 변수 (Outcome Variables) 등으로 구별할 수 있습니다.
임상시험에서는 유효성 평가와 관련될 결과변수 이외에도 타 변수 데이터에 대해서도 충실히 수집되어야만 임상시험용의약품에 대한 적절하고 종합적인 평가가 가능하며, 임상시험에서 획득되는 데이터 하나하나의 소중함을 알고 임상시험에 임해야 할 것으로 생각됩니다.
예컨데 유효성 평가의 경우, 환자집단의 특성에 대해 인구통계학적인 특성, 질병상태, 그 외 중요하다고 생각되는 공변량 등이 유효성 평가 시 고려되어야 하는데 이들은 기저치 변수에 해당합니다. 또한 임상시험에서 중도탈락된 환자들에 대한 정보 등은 공정변수에 해당합니다. 상기 언급한 데이터들은 임상시험 분석의 통계방법과 시험결과의 해석에 영향을 미칠 수 있는 요인들이며, 유효성 분석 및 평가 시 종합적으로 고려되어야 할 사항들입니다.
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 신약개발의 프로세스
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 신약 후보물질의 발굴
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 신약 후보물질의 생산과 품질관리
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 완제 및 원료의약품 GMP
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 비임상 효력시험
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 일반약리시험과 안전성약리시험
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 흡수분포대사배설시험 및 약물상호작용시험
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 독성시험 (비임상 안전성평가시험)
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 비임상시험관리기준 (GLP)
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 비임상시험과 임상시험의 연계
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 임상개발 개요
[보건의료/신약개발 과정의 이해] - 후보 물질 발굴과 비임상시험 | 임상시험의 과학적 측면

댓글